eSignature Legality for Drug Testing Consent Agreement in European Union
- Quick to start
- Easy-to-use
- 24/7 support






Award-winning eSignature solution




Simplified document journeys for small teams and individuals




We spread the word about digital transformation










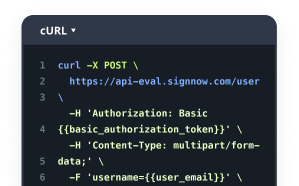
Why choose airSlate SignNow
-
Free 7-day trial. Choose the plan you need and try it risk-free.
-
Honest pricing for full-featured plans. airSlate SignNow offers subscription plans with no overages or hidden fees at renewal.
-
Enterprise-grade security. airSlate SignNow helps you comply with global security standards.







Your complete how-to guide - esignature legality for drug testing consent agreement in european union
eSignature legality for Drug Testing Consent Agreement in European Union
When it comes to ensuring the legality of drug testing consent agreements in the European Union, having an eSignature solution like airSlate SignNow can streamline the process. With airSlate SignNow, businesses can easily send and eSign documents while ensuring compliance with EU regulations.
How to Use airSlate SignNow for eSigning Documents:
- Launch the airSlate SignNow web page in your browser.
- Sign up for a free trial or log in.
- Upload a document you want to sign or send for signing.
- If you're going to reuse your document later, turn it into a template.
- Open your file and make edits: add fillable fields or insert information.
- Sign your document and add signature fields for the recipients.
- Click Continue to set up and send an eSignature invite.
airSlate SignNow empowers businesses to send and eSign documents with an easy-to-use, cost-effective solution. It offers great ROI with a rich feature set, it's easy to use and scale for SMBs and Mid-Market, has transparent pricing without hidden fees, and provides superior 24/7 support for all paid plans.
Experience the benefits of airSlate SignNow today and streamline your document signing process!
How it works
Rate your experience
Understanding the eSignature Legality for Drug Testing Consent Agreements in the European Union
The eSignature legality for drug testing consent agreements within the European Union is governed by the eIDAS Regulation, which establishes a legal framework for electronic signatures. This regulation ensures that electronic signatures hold the same legal weight as handwritten signatures, provided they meet specific criteria. In the context of drug testing consent agreements, this means that employers can obtain consent from employees electronically, streamlining the process while maintaining compliance with legal standards.
How to Use the eSignature Legality for Drug Testing Consent Agreements
To use the eSignature legality for drug testing consent agreements, businesses should first ensure that their electronic signature solution complies with the eIDAS Regulation. Once compliance is verified, employers can create a digital consent form that includes all necessary information regarding the drug testing process. Employees can then fill out this form online, providing their consent with an electronic signature. This method not only simplifies the process but also allows for easy storage and retrieval of signed documents.
Steps to Complete the eSignature Legality for Drug Testing Consent Agreements
Completing a drug testing consent agreement electronically involves several key steps:
- Prepare the consent form, ensuring it includes all relevant details such as the purpose of the drug test and the employee's rights.
- Upload the document to an eSignature platform like airSlate SignNow.
- Invite the employee to review and sign the document by sending it for signature.
- The employee fills out the required information and applies their electronic signature.
- Once signed, the document is securely stored and can be accessed as needed.
Key Elements of the eSignature Legality for Drug Testing Consent Agreements
When drafting a drug testing consent agreement for electronic signing, several key elements must be included:
- Clear identification of the parties involved.
- A detailed description of the drug testing process.
- Information on how the test results will be used and shared.
- Explicit consent from the employee, acknowledging their understanding of the process.
Security & Compliance Guidelines
Ensuring security and compliance when using eSignatures for drug testing consent agreements is critical. Organizations should implement the following guidelines:
- Use a reputable eSignature platform that complies with eIDAS and other relevant regulations.
- Ensure that all documents are encrypted during transmission and storage.
- Maintain an audit trail that records all actions taken on the document, including when it was sent, viewed, and signed.
- Regularly review and update security measures to protect sensitive information.
Digital vs. Paper-Based Signing
Choosing between digital and paper-based signing for drug testing consent agreements can significantly impact efficiency. Digital signing offers numerous advantages:
- Faster processing times, as documents can be signed and returned instantly.
- Reduced paper usage, contributing to sustainability efforts.
- Improved tracking and management of signed documents through electronic storage solutions.
-
Best ROI. Our customers achieve an average 7x ROI within the first six months.
-
Scales with your use cases. From SMBs to mid-market, airSlate SignNow delivers results for businesses of all sizes.
-
Intuitive UI and API. Sign and send documents from your apps in minutes.
FAQs
-
What is the esignature legality for drug testing consent agreement in European Union?
The esignature legality for drug testing consent agreement in European Union is governed by the eIDAS Regulation, which recognizes electronic signatures as legally binding. This means that businesses can use airSlate SignNow to obtain consent for drug testing electronically, ensuring compliance with EU regulations.
-
How does airSlate SignNow ensure compliance with esignature legality for drug testing consent agreements?
airSlate SignNow adheres to the eIDAS Regulation, ensuring that all electronic signatures are secure and legally recognized. Our platform provides features such as audit trails and identity verification, which are essential for maintaining the esignature legality for drug testing consent agreement in European Union.
-
What features does airSlate SignNow offer for managing drug testing consent agreements?
airSlate SignNow offers a range of features including customizable templates, secure storage, and real-time tracking of document status. These features enhance the efficiency of managing drug testing consent agreements while ensuring esignature legality for drug testing consent agreement in European Union.
-
Is there a cost associated with using airSlate SignNow for drug testing consent agreements?
Yes, airSlate SignNow offers various pricing plans to accommodate different business needs. Each plan provides access to features that support the esignature legality for drug testing consent agreement in European Union, making it a cost-effective solution for businesses.
-
Can airSlate SignNow integrate with other software for drug testing processes?
Absolutely! airSlate SignNow integrates seamlessly with various software applications, enhancing your workflow for drug testing processes. This integration supports the esignature legality for drug testing consent agreement in European Union by streamlining document management.
-
What are the benefits of using airSlate SignNow for drug testing consent agreements?
Using airSlate SignNow for drug testing consent agreements offers numerous benefits, including increased efficiency, reduced paperwork, and enhanced security. By ensuring esignature legality for drug testing consent agreement in European Union, businesses can confidently manage their compliance needs.
-
How secure is airSlate SignNow for handling sensitive drug testing consent agreements?
airSlate SignNow employs advanced security measures, including encryption and secure access controls, to protect sensitive information. This commitment to security ensures the esignature legality for drug testing consent agreement in European Union, allowing businesses to handle documents safely.






Related searches to esignature legality for drug testing consent agreement in european union
Join over 28 million airSlate SignNow users
Get more for esignature legality for drug testing consent agreement in european union
- Unlocking the power of online signature legitimacy for ...
- Unlock Online Signature Legitimateness for Business ...
- Enhance Business Partnerships with Legitimate Online ...
- Unlock the Power of Online Signature Legitimateness for ...
- Boost Your Business with Online Signature ...
- Unlock Legitimate Online Signature Solutions for ...
- Online Signature Legitimateness for Business Purchase ...
- Unlock the Power of Online Signature Legitimateness for ...