Force Validated Field with airSlate SignNow
Award-winning eSignature solution




Do more on the web with a globally-trusted eSignature platform
Standout signing experience
Trusted reports and analytics
Mobile eSigning in person and remotely
Industry polices and compliance
Force validated field, faster than ever
Useful eSignature add-ons
See airSlate SignNow eSignatures in action




airSlate SignNow solutions for better efficiency
Our user reviews speak for themselves









Why choose airSlate SignNow
-
Free 7-day trial. Choose the plan you need and try it risk-free.
-
Honest pricing for full-featured plans. airSlate SignNow offers subscription plans with no overages or hidden fees at renewal.
-
Enterprise-grade security. airSlate SignNow helps you comply with global security standards.

Your step-by-step guide — force validated field
Using airSlate SignNow’s electronic signature any company can speed up signature workflows and eSign in real-time, supplying an improved experience to customers and employees. force validated field in a few simple actions. Our mobile apps make work on the run feasible, even while offline! Sign documents from anywhere in the world and make deals quicker.
Follow the stepwise guideline to force validated field:
- Log on to your airSlate SignNow profile.
- Locate your record within your folders or import a new one.
- Open up the template adjust using the Tools list.
- Drag & drop fillable boxes, add textual content and sign it.
- List several signers by emails configure the signing order.
- Specify which individuals will receive an completed copy.
- Use Advanced Options to restrict access to the record add an expiry date.
- Tap Save and Close when done.
Moreover, there are more innovative features available to force validated field. List users to your shared digital workplace, browse teams, and track collaboration. Millions of consumers all over the US and Europe agree that a solution that brings people together in one cohesive digital location, is what enterprises need to keep workflows working easily. The airSlate SignNow REST API enables you to embed eSignatures into your application, internet site, CRM or cloud storage. Check out airSlate SignNow and get quicker, smoother and overall more effective eSignature workflows!
How it works
airSlate SignNow features that users love






See exceptional results force validated field with airSlate SignNow
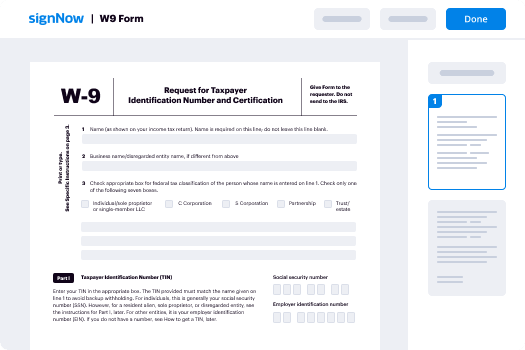
How to complete and sign a PDF online
Try out the fastest way to force validated field. Avoid paper-based workflows and manage documents right from airSlate SignNow. Complete and share your forms from the office or seamlessly work on-the-go. No installation or additional software required. All features are available online, just go to signnow.com and create your own eSignature flow.
A brief guide on how to force validated field in minutes
- Create an airSlate SignNow account (if you haven’t registered yet) or log in using your Google or Facebook.
- Click Upload and select one of your documents.
- Use the My Signature tool to create your unique signature.
- Turn the document into a dynamic PDF with fillable fields.
- Fill out your new form and click Done.
Once finished, send an invite to sign to multiple recipients. Get an enforceable contract in minutes using any device. Explore more features for making professional PDFs; add fillable fields force validated field and collaborate in teams. The eSignature solution gives a secure process and operates based on SOC 2 Type II Certification. Ensure that your data are protected and that no person can take them.
How to eSign a PDF file in Google Chrome
Are you looking for a solution to force validated field directly from Chrome? The airSlate SignNow extension for Google is here to help. Find a document and right from your browser easily open it in the editor. Add fillable fields for text and signature. Sign the PDF and share it safely according to GDPR, SOC 2 Type II Certification and more.
Using this brief how-to guide below, expand your eSignature workflow into Google and force validated field:
- Go to the Chrome web store and find the airSlate SignNow extension.
- Click Add to Chrome.
- Log in to your account or register a new one.
- Upload a document and click Open in airSlate SignNow.
- Modify the document.
- Sign the PDF using the My Signature tool.
- Click Done to save your edits.
- Invite other participants to sign by clicking Invite to Sign and selecting their emails/names.
Create a signature that’s built in to your workflow to force validated field and get PDFs eSigned in minutes. Say goodbye to the piles of papers on your desk and start saving money and time for additional crucial activities. Picking out the airSlate SignNow Google extension is a great convenient choice with plenty of benefits.

How to eSign an attachment in Gmail
If you’re like most, you’re used to downloading the attachments you get, printing them out and then signing them, right? Well, we have good news for you. Signing documents in your inbox just got a lot easier. The airSlate SignNow add-on for Gmail allows you to force validated field without leaving your mailbox. Do everything you need; add fillable fields and send signing requests in clicks.
How to force validated field in Gmail:
- Find airSlate SignNow for Gmail in the G Suite Marketplace and click Install.
- Log in to your airSlate SignNow account or create a new one.
- Open up your email with the PDF you need to sign.
- Click Upload to save the document to your airSlate SignNow account.
- Click Open document to open the editor.
- Sign the PDF using My Signature.
- Send a signing request to the other participants with the Send to Sign button.
- Enter their email and press OK.
As a result, the other participants will receive notifications telling them to sign the document. No need to download the PDF file over and over again, just force validated field in clicks. This add-one is suitable for those who like concentrating on more important aims instead of burning up time for nothing. Enhance your daily monotonous tasks with the award-winning eSignature solution.


How to sign a PDF on the go with no application
For many products, getting deals done on the go means installing an app on your phone. We’re happy to say at airSlate SignNow we’ve made singing on the go faster and easier by eliminating the need for a mobile app. To eSign, open your browser (any mobile browser) and get direct access to airSlate SignNow and all its powerful eSignature tools. Edit docs, force validated field and more. No installation or additional software required. Close your deal from anywhere.
Take a look at our step-by-step instructions that teach you how to force validated field.
- Open your browser and go to signnow.com.
- Log in or register a new account.
- Upload or open the document you want to edit.
- Add fillable fields for text, signature and date.
- Draw, type or upload your signature.
- Click Save and Close.
- Click Invite to Sign and enter a recipient’s email if you need others to sign the PDF.
Working on mobile is no different than on a desktop: create a reusable template, force validated field and manage the flow as you would normally. In a couple of clicks, get an enforceable contract that you can download to your device and send to others. Yet, if you want a software, download the airSlate SignNow mobile app. It’s secure, quick and has an excellent layout. Enjoy seamless eSignature workflows from the business office, in a taxi or on a plane.

How to sign a PDF employing an iPhone
iOS is a very popular operating system packed with native tools. It allows you to sign and edit PDFs using Preview without any additional software. However, as great as Apple’s solution is, it doesn't provide any automation. Enhance your iPhone’s capabilities by taking advantage of the airSlate SignNow app. Utilize your iPhone or iPad to force validated field and more. Introduce eSignature automation to your mobile workflow.
Signing on an iPhone has never been easier:
- Find the airSlate SignNow app in the AppStore and install it.
- Create a new account or log in with your Facebook or Google.
- Click Plus and upload the PDF file you want to sign.
- Tap on the document where you want to insert your signature.
- Explore other features: add fillable fields or force validated field.
- Use the Save button to apply the changes.
- Share your documents via email or a singing link.
Make a professional PDFs right from your airSlate SignNow app. Get the most out of your time and work from anywhere; at home, in the office, on a bus or plane, and even at the beach. Manage an entire record workflow effortlessly: generate reusable templates, force validated field and work on PDF files with business partners. Transform your device into a powerful business tool for executing offers.

How to eSign a PDF file Android
For Android users to manage documents from their phone, they have to install additional software. The Play Market is vast and plump with options, so finding a good application isn’t too hard if you have time to browse through hundreds of apps. To save time and prevent frustration, we suggest airSlate SignNow for Android. Store and edit documents, create signing roles, and even force validated field.
The 9 simple steps to optimizing your mobile workflow:
- Open the app.
- Log in using your Facebook or Google accounts or register if you haven’t authorized already.
- Click on + to add a new document using your camera, internal or cloud storages.
- Tap anywhere on your PDF and insert your eSignature.
- Click OK to confirm and sign.
- Try more editing features; add images, force validated field, create a reusable template, etc.
- Click Save to apply changes once you finish.
- Download the PDF or share it via email.
- Use the Invite to sign function if you want to set & send a signing order to recipients.
Turn the mundane and routine into easy and smooth with the airSlate SignNow app for Android. Sign and send documents for signature from any place you’re connected to the internet. Generate professional-looking PDFs and force validated field with just a few clicks. Come up with a flawless eSignature process with only your smartphone and increase your general efficiency.
Get legally-binding signatures now!
What active users are saying — force validated field








Force validated field
okay so without any further ado let's talk a little bit about force fields okay I'll give you a brief introduction to force fields and and my and my research which is involved with force field optimization so there if there really is a very there's a vast range of molecular simulation methods and in this open mm workshop we are mainly concerned with the routing atomistic classical molecular mechanics using empirical force fields right but you can see that in the big picture there's really a very wide range of ways to simulate atoms and molecules using you can you can use a very level of detail and run what are called coarse-grained simulations in which the particles in your simulations don't represent atoms at all they really just they really represent collections of atoms so an entire amino acid can be represented using one particle and that allows you to simulate say entire viruses or even cells but at a much lower level of detail and to go to a higher level of detail you have methods that tree feel that treat the electrons explicitly like the hartree-fock method or density functional theory which are all approximations to sheron's equations which can only be solved for systems that contain one electron okay and and in general the more physical detail you include into your simulation the more costly the simulation is going to be but more physical detail doesn't always mean a more accurate model right because there are many definitions for accuracy and we are always searching for the simplest essential description of our system because that is going to enable us to run rapid and accurate simulations so that's kind of like our end goal in designing good force fields okay so these are the different types of interactions that are available in a typical force field you have the stretching of harmonic bonds the bending of of angles between three atoms the rotation of a dihedral angle and also interactions between non bonded atoms which usually take the form of a electrostatic interaction plus a Vander Waals interaction as you can see if this is a sodium ion and a chloride ion they attract each other with an electrostatic interaction and when they get close enough they repel and the accuracy of your simulation depends critically on your choice of the parameters because there really is no underlying sure turns of assuring there's equation - approximately right you can really write down any equation you want and your simulation can do anything if you wanted to reflect reality you better choose a good set of parameters so what set of parameters do you choose there have been groups that have developed force fields for a very long time there's a relatively small number of these groups and they've published a number of force fields over the years for example the Amber consortium has developed a series of amber force fields at large like FF 94 96 99 oh three and and ten and there's also various modifications to the dihedral angles and ff99 that give you better protein structures there's a generalized amber force field which is more of a procedure that allows you to build to build a force field for any small molecule that is consistent with the rest of the invar force field okay and that's just for amber there's also there's o pls and charm and amoeba and and even though there's a relatively small number of research groups making these force fields to you when when you're starting out on a research project which one do you choose you know there's a lot of force fields for you to choose from and the amount of validation in the literature is kind of limited with the exception of a very small number of force fields that a lot of people run like a lot of simulations on similarly for water there are many force fields that describe water the most well-known ones are tip free Pete at 4:00 p.m. tip 5 P where the stands are transferable intermolecular potential and and perhaps a little bit ironically most of these protein force fields are developed in conjunction with the tip 3p water model which also happens to be probably the worst water model that I've listed on there like for example when people had simulated the melting point and boiling point of tip 3p water they find that it melts at a minus 150 degrees Celsius and boils at night- 90 degrees Celsius which you know it's not what water really does do you really want that interacting with your protein you know well I'm not sure probably not so there's way too many force fields to choose from and and this is probably becoming a headache for a lot of people who are doing protein simulations if you're if your result doesn't agree with the experiment this is a this is a possible source of error okay and and it might lead some people to ask the question can we create a force field that is best for our particular research project I think there are three main steps to make to creating a force field I mean of course force field development is a very very discipline everyone does a little bit differently but probably most of probably most force field development approaches have these three main steps the first is to choose a functional form okay and I gave this amoeba sly I gave the slide yesterday and my amoeba presentation but the main reason why this slide is here is that there are different ways to describe the electrostatic interactions for example the amber force field just uses point partial charges that are located on the atoms whereas amoeba uses a much more complex description with a second-order multipole expansion on each atom plus putting a polarizable dipole on each of these atoms the amoeba description is more accurate but it is also more costly so it's really up to you to choose what level of what level of detail you want for the electrostatic description and all the other interactions okay the second step is to choose a set of reference data the reference data as the is the data that you want your force field to reproduce and hopefully the reference data is very well is something that can reflect reality for instance you can choose your reference data to come from a high accuracy quantum mechanical calculation this is what a 2d potential energy scan of the dihedral angles of a molecule might look like and when you scan these angles and compute the energies using say a really high-level theory like MP 2 or CCSD or something it might take you a few days but you'll end up with a potential energy surface like this one that you would like your force field to be able to reproduce you can also do something like compute the electrostatic potential from a quantum method on a molecular surface you can see I have a plot of the electrostatic potential on a density isosurface where I think blue is negative and red is positive and you might want the charges in your force field to reproduce that and that's how the RESPA method works and the generalized amber force field and and why not have the force field also reproduce some some measurements from experiment because that's what we're often trying to compare to experiment and where and maybe if we have a really good force field we can try to predict experimental results right so you so you can see that it's possible to do something like simulated EMA an NMR chemical shift with with the molecular dynamics simulation as long as you have a spectroscopic model that takes your simulation trajectory to the predicted spectra and this grid here has various protein force fields on the x-axis and various water models on the y-axis and the and the color indicates how well the combination of protein force field and water force field reproduces the NMR chemical shifts for 500 different peptides and as you can see the force fields that are specifically parametrized to get these chemical shifts right do the best those are the ones in blue where I say F F 96 combined with and implicit solvent modeled GPS a really doesn't do very well so these can be used to validate forcefields but my main point here is that they can also be used to optimize your force field this the third step is to construct an objective function and apply an optimization method to minimize it the objective function is a function of your force field parameters okay it might take very long to evaluate but essentially it's supposed to measure the disagreement between the reference data and the corresponding simulation result so the better your force field the smaller the objective function and the optimization algorithm is going to be a strategy for choosing different parameters to to go into the objective function because well the mean the main point here is that you can't evaluate the objective function for every possible combination of parameters the objective function is probably a very complicated function you're not going to know its global minimum so rather you need to apply a numerical optimization algorithms who search for the minimum if you have a very small number of parameters like 1 or 2 you can apply a grid scan which really goes through the entire parameter space and some kind of finite domain but grid scans are not are not feasible if you have large numbers of parameters because the number of a grid points is exponential right you can also use derivative based optimization algorithms in which you evaluate the objective function at an initial guess of your parameters but you also evaluate its derivative optionally you can also evaluate the second derivative and that tells you the direction and also how far you should step so if it's so if I'm so if I'm currently here and I've evaluated the derivative it will tell me that if I go in this direction the objective function will go down the second derivative tells me that the surface will curve in this direction so I will step in this direction and as you can see I will end up moving towards the minimum the third method is using a stochastic or some say global optimization methods like simulated annealing or genetic algorithms these methods really are kind of like got a guided random search so so simulated annealing essentially performs some kind of it performs something like a Monte Carlo simulation or molecular dynamics simulation but in your parameter space and the parameters are given random perturbations but they also prefer to go where the objective function is low so if you're really worried about derivative based optimizations finding local minima okay you can apply these global methods to hop over barriers but just keep in mind that the number of objective function evaluations is generally higher using these stochastic methods okay so these are the concepts and I'll talk a little bit about my implementation I wrote a program called force balance to to optimize force fields this is just a logo of the program and if you're wondering the Chinese character in the middle means balance okay there's a direct interface with open mm so if you like using open mm force balance is very easy to use for parametrizing a force field it's highly flexible and easily easily extensible and also freely available if you go to Cintiq a org which is the website you've been going to to get all of the downloads for this workshop you can also go to the force balance page where you can download the code here's kind of how the code is laid out there are three main concepts that are that are represented by classes I was wondering can you see it because it's kind of bright you can see it okay so I send that you want to pick a functional form that's mainly contained in the force field class the objective function classes will will basically evaluate this objective function which is how well your force field is reproducing the reference data and the optimizer will contain all the optimization algorithms that allow you to do a perimeter search and all of the and all of the specific types of force fields reference data and optimization methods are really just subclasses of these three main classes and there's an in addition there's a lot of features in here that that that basically make this optimization problem well will basically make it easier for you to do the specific problem of force field optimization for example while force balance doesn't actually contain any equations to evaluate these interactions rather it operates through interfaces - - you know mature and well validated programs like open mmm grow max and and so on so all of the simulation results is actually getting from these programs and it's reading in the reference data to construct the objective function and then it applies these optimization methods to to tune the parameters okay so now for a simple application I talked about the ameba force field yesterday and that and i and i mentioned that it's a that is one of the more accurate force fields that are out there it's a polarizable force field right and via and the foundation of any biomolecular force field is a good water model now the ameba water model was published in 2003 and it's still regarded as one of the most accurate water models out there however it's very slow right and then and one of the reasons why it's really slow is that can it contains these things that are called mutually induced dipoles it's because when is because you have B polarizable dipoles and imagine that every simulation step your polarizable dipoles are going to feel the influence of the electric field from your static multi-pulse okay so that means the polarizable dipoles show up but once they show up they start to influence each other so the precise values of the polarizable dipoles need to be solved self consistently in many iterations at every simulation step that's very costly and in fact if you rewrite your equations in a funny way where the where the polarizable dipoles don't interact with each other okay they if they only interact with the static multi-pulse but you don't need to do the self consistent solution anymore and that will reduce the computational cost by two types of five times depending on the implementation and open mmm it's five times faster okay in fact you could just there's an option for creating the system when you specify polarization direct and then it will allow you to use the ameba force field but without any interactions between polarizable dipoles but the reason why didn't talk about this is because if you set polarization direct the equations become very different and the results get a lot worse if you if you take a look at via the simulated density of water using the amoeba force field you see the red curve here you can see that around room temperature 25 degrees Celsius it's in a it's in good agreement with experiment and also at higher temperatures but at lower temperatures the disagreement is pretty big and you and in the insight you also see that this is the enthalpy of vaporization of water with respect to temperature the the ameba force field gets the slope of the curve a little bit high but at room temperature the agreement is generally good now if you run polarization direct and you try to calculate the density and enthalpy of vaporization curves everything is qualitatively wrong you see the density does not have a density maximum it's well known that water has a density maximum around four degrees in this temperature range the direct model does not have a gonna city maximum and furthermore the enthalpy of vaporization is too low you can also compare to some theoretical data if you look at the if you run a bunch of calculations on little water clusters using the mp2 method and look at the scatter plot of energies pudding say if you put a quantum of x-axis and the force field on the y-axis the original ameba force field which is in the upper right gives a pretty good agreement whereas the polarization direct model gives an agreement that significantly worse because the scatter is bigger so this is a good application for us there's a about nineteen tunable parameters in the in this force field and we want to see if we use polarization direct is it possible to recover the accuracy of the original ameba force field which will give us a five times speed up and hopefully still give us a physically reasonable picture of the polarization so this is what we get when we after we run the optimization so you see that there's a new blue curve on there the blue curve is polarization direct but we've applied force balance to optimize the parameters okay and you can and you can see that not only do we get the density of water correct around room temperature but we also get the density to be correct with them the entire temper arranged and the temperature of maximum density is now in the right place the enthalpy of vaporization is also brought back into agreement with the original ameba force field there used to be too low way down here now it's up here the slope is still a little bit high and you can see where or before or polarization direct had a large scatter with respect to the quantum energies and forces now now the scatter is better and all these are the properties that we fitted to and and you say well well it's obvious that if you do curve fitting I mean this is really glorified curve fitting right that the results are going to be better and the true test of the force field is whether is whether it can predict properties that were not in the objective function and that's a and I think that's completely correct so when we run some we run some simulations of other properties that we did not include in our objective function we also find the results to be quite good like that like the diffusion constant is closer to experiment than the original amoeba the dielectric constant is still on the right place and the temperature of maximum density which we technically didn't fit to directly is also at a good number if you look at the radial distribution function of two oxygen atoms in liquid water it's also pretty close to the experiment so we do think that using for spells we created a good water force field that's competitive with the best ones that are out there so how is force balance integrated with open mm in another one of these little diagrams I seem to enjoy creating these the blue diamonds represent things like concepts that are within force balance and the yellow corresponds to things that are inside open in them and because force balance is entirely written in Python it integrates pretty seamlessly with open mm basically whenever whenever I want to use open mm to evaluate say the energy and the force for a particular configuration or if I want it to simulate an ensemble average property which then goes into my objective function I create the system execute the simulation save the data and then it comes back into the objective function it's very simple and it's very natural and in fact open atom has been very helpful for bringing force balance aware currently is now I don't have a demo for forced balance even though like I could have technically had one but but I'm gonna mention briefly how you would use it if you want it to use it the parameters to be optimized are specified by labeling the force field XML file so if you came to my breakout session yesterday you know that the you know that the force field XML file specifies all the interactions that that you have when you set up your system and for example this Clause is the ameba harmonic bond force which is really just a harmonic interaction between two atoms with some an harmonic Corrections and what I've done in the XML element is to add an attribute parameterize equals length comma K and when you and the force balance will read this attribute open and M doesn't touch it and and in those that this number is a number that needs to be optimized and this number needs to be optimized so as force balance runs it will write new XML files with these numbers replaced by the optimized valleys at each optimization step will write new parameter files and there are several force field formats that are supported for example the grow max ITP format and and so on but it's really most natural to do this in an open mmm and other thing that you might find nifty is that well you often don't want parameters to be completely independent variables like for example I don't want to optimize all the charges because they might not add to 0 anymore right so you can actually you can actually write XML in such a way that any force field parameter here is an arbitrary mathematical function of an independent parameter so so it's really quite versatile and and hopefully written in such a way that is that is natural for general force field optimization problems and not just mine this is a force balance input file it's a I've modeled it after the Q chem input file because I've used Q Ken the quantum chemistry program a lot but you can see there's basically a set there's basically a global options section that specifies options for the forcefield and for the optimization algorithm and for the different components of your objective function while every component of the objective function is a simulation and a corresponding set of reference data and so every one of those gets a gets a clause like this so this is a there's an example input file the real and profile might be longer because force balance does have many options I have an instruction male and a doxygen page that you can they can consult to get information on all of the options just by going to the Cintiq a project page unlike open mm I guess force balance is not designed to be to be a library that you import modules from and do anything in maybe in the future it could be like that but for now we just have a script called force balance that PI that you just run and it does what you want so so you run the script it does the optimization I mean if you if you want you could probably like load the various libraries to do various things but this kind of ties all of it together furthermore because optimizations are often multi-day tasks and you might want to tweak it and restart you can really paste sections of the output back into the input and it will pick up where you left off okay one last little detail of of things we put into force balance is that we put very strict regularization now regularization means that you don't allow your optimization to go too far because your objective function is not going to be a complete description of reality right it's only going to contain certain parts of your reference data and it's possible to overfit it if you over fit it they end up with unphysical parameter values and when you go off to simulate something that wasn't in your objective function that you do horribly so so the way we take care of that is really a is really very simple you add a penalty function on to your original objective function that make sure that makes sure your parameters don't go too far away from where you started them from so you start them from some physically reasonable guess and the idea is that your optimization won't move too far from there although the precise valley of your initial guess is not going to be so important it's just important that they're physically reasonable the blue line you see here might be the original objective function and if I fit it I might I might find the parameter ends up here and who knows this might be a this might be the angle of a bond and and I've ended up with the angle of the bond being 15 degrees smaller that might be unphysical right in that case you might want to add a penalty function which actually has a strict correspondence in a Bayesian probability theory if you if you just put a parabola on the objective function that's called a Gaussian prior and that means that when you do the optimization your parameter is not going to move so far okay you can also apply something called a laplacian prior which is which where you basically add the absolute value of the parameter and not look and not like the parameters squared onto the objective function and the unique feature of using a laplacian prior is that some parameters are not going to change at all while other parameters are optimized and you don't need to you don't need to specify which ones which parameters don't change it which ones change the algorithm kind of picks them out for you okay so so we so we hope that force balance will allow a force field optimization to be more systematic so it might like we if you use this method you don't need to choose parameters by hand anymore however that's but it's not to say that this program is a replacement for a scientific decision making because we all make decisions that go into the force field it's just that we're not picking the precise values of the parameters rather we are choosing what types of data go into our objective function and how much the parameters are allowed to vary so the scientific decision-making is done at a much higher and I would also venture to say more scientific level and furthermore because well because this program really allows you to optimize a force field in a way that's similar to running a molecular mechanics simulation so instead of a year-long project that's very that's not reproducible because you're doing you know a very like a varus collection of things here you just write in put file is set up the calculation and you run it and that might improve the reproducibility of force field development and furthermore because because every force balance calculation is kind of self-contained it contains all the contains all the reference data and optimization algorithms we hope that this would give everybody the infrastructure for making good force fields for example if you have uncommon molecules even like not mainstream not of proteins so if you're so if you're a so if you're a hipster basically you know there might not be a force field that is optimized for you and you can use this method to improve the accuracy of your simulation where force field development efforts are relatively sparse and force balance is all inclusive in the sense that it's really easy to write interfaces with it with any simulation software because if I I knew that if I wanted this software to be general it has to interface with with anyone's anybody's simulation software of choice so interfaces are very easy to write they're a little bit like plugins and in general it reduces the headache of Forestville development and allows us to focus on a science so I'd be happy to talk to you talk to you more about this later but but thank you
Show moreFrequently asked questions
How do I add an electronic signature to a PDF in Google Chrome?
How do I eSign a PDF on a PC?
How can I have someone sign on a PDF file?
Get more for force validated field with airSlate SignNow
- ESignature TXT
- Prove electronically signed Camping Trip Itinerary
- Endorse digisign Privacy Policy
- Authorize electronically sign Video Proposal Template
- Anneal mark Travel Planning Registration
- Justify esign Office Supplies Inventory
- Try countersign Boat Slip Lease Agreement
- Add Joint Venture Agreement signed electronically
- Send Wedding Photography Contract Template electronically sign
- Fax Sorority Recommendation Letter Template countersignature
- Seal Management Report mark
- Password 1040 Form signed
- Pass Leave of Absence Letter digi-sign
- Renew Snow Removal Contract digital sign
- Test Rent Receipt initial
- Require Tripartite Agreement Template signature
- Print receiver autograph
- Champion renter eSignature
- Call for collector eSign
- Void Non-Disclosure Agreement (NDA) template sign
- Adopt Assumption Agreement template electronically signing
- Vouch Fundraiser Ticket template mark
- Establish Silent Auction Gift Certificate template eSignature
- Clear Grant Proposal Template template autograph
- Complete Maintenance Work Order template digital sign
- Force Web Design Proposal Template template electronic signature
- Permit Basic Employment Resume template signed electronically
- Customize Power of Attorney template electronically sign